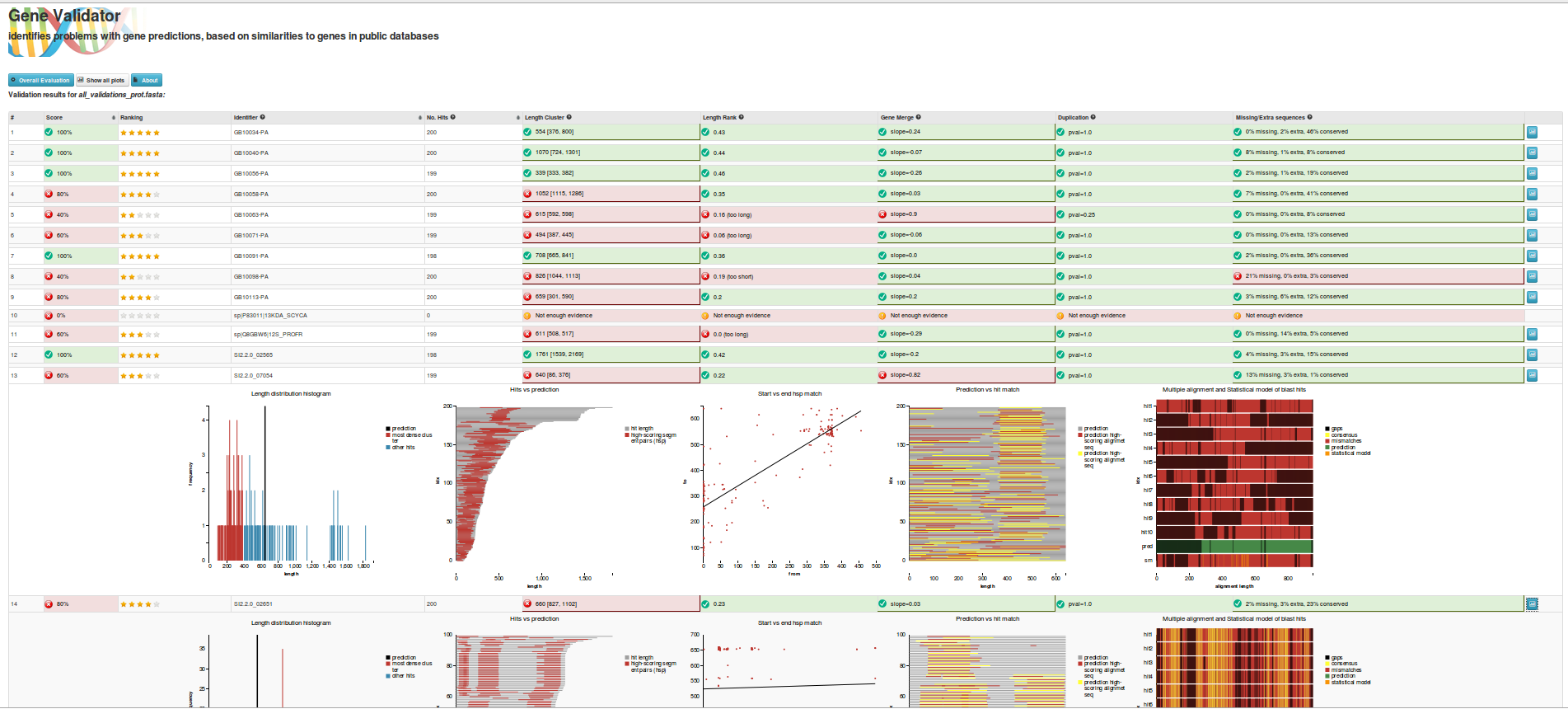
Identify problems with predicted genes
This is a NESCent project, funded by Google during Google Summer of Code 2013 and mentored by Anurag Priyam and Yannick Wurm.
Authors
- GSoC student: Monica Dragan
- Mentors: Anurag Priyam and Yannick Wurm
Resources
Summary
1) Length Validation via clusterization
2) Length Validation via ranking
5) Validation based on multiple alignment - for proteins only
6) Blast Reading Frame Validation - for nucleotides only
7) Open reading frame - for nucleotides only
8) Codon usage - this is future work
Add the new validation to the tool
A. Project description
Genome sequencing is now possible at almost no cost. However, obtaining accurate gene predictions remains a target hard to achieve with the existing biotechnology.
We designed a tool that helps identifying problems with gene predictions, based on similarities with data from public databases (like Swissprot and Uniprot). We apply a set of validation tests that provide useful information about the problems that appear in the predictions, in order to make evidence about how the gene curation can be made or whether a certain predicted gene may not be considered in other analysis.
Our main target users are the biologists, who have large amounts of genetic data that has to be manually curated, and our tool will prioritize the genes that need to be checked and suggest possible error causes.
B. Validations
In order to highlight the problems that appear in the predictions and suggest possible error causes, we developed 7 validation test:
1) Length validation via clusterization
2) Length validation via ranking
3) Duplication
4) Gene merge
5) Multiple alignment
6) Blast Reading Frame - applicable only for nucleotide sequences
7) Open reading frame - applicable only for nucleotide sequences
8) Codon usage -- under development
Data needed for each validation is retrieved from the BLAST output and used as follows (aliases and tags are those used in BLAST):
Blast param | sseqid | sacc | slen | qstart | qend | sstart | send | length | qseq | sseq | qframe | evalue | Query raw_seq | Hit raw_seq |
Valid-ation | ||||||||||||||
lenc | * | x | * | |||||||||||
lenr | * | x | * | |||||||||||
frame | * | x | * | |||||||||||
merge | * | x | x | x | x | * | ||||||||
dup | * | x | x | x | x | x | x | * | ||||||
orf | * | ~~ | * | x | ||||||||||
align | * | x | ~~ | * | x | |||||||||
codon | * | ~~ | * | x |
x = mandatory parameter
~~ = May be needed
* = Nice to have
Each validation is described further in this section.
1) Length Validation via clusterization
Error causes
|
Input data
|
Class information:
|
WorkflowAim : we are interested to find out if the length of the predicted sequence belongs to the distribution of the hit lengths (in other words, how close is the length of the prediction to the majority of the lengths of the hits)
(1) Our approach to find the majority of lengths among the reference lengths is by a typical hierarchical clusterization:
(2)
(3) (4) |
Code#clusterization by length contents = lst.map{ |x| x.length_protein.to_i }.sort{|a,b| a<=>b} hc = HierarchicalClusterization.new(contents) clusters = hc.hierarchical_clusterization max_density = 0; max_density_cluster_idx = 0; clusters.each_with_index do |item, i| if item.density > max_density max_density = item.density max_density_cluster_idx = i; end end clusterization = [clusters, max_density_cluster_idx] @clusters = clusterization[0] @max_density_cluster = clusterization[1] limits = @clusters[@max_density_cluster].get_limits prediction_len = @prediction.length_protein @validation_report = LengthClusterValidationOutput.new(prediction_len, limits) plot1 = plot_histo_clusters @validation_report.plot_files.push(plot1) plot2 = plot_len_clusters @validation_report.plot_files.push(plot2) |
Plots
(3) (4)
|
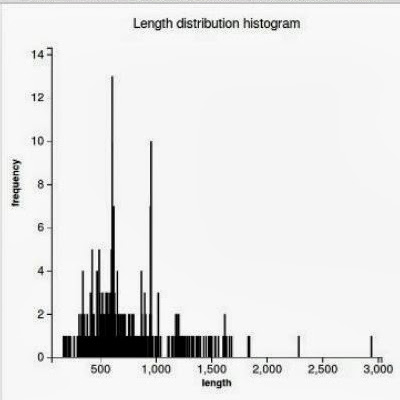
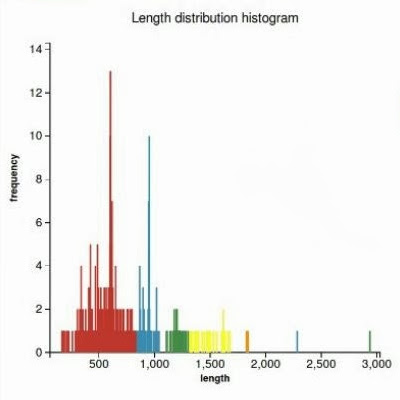
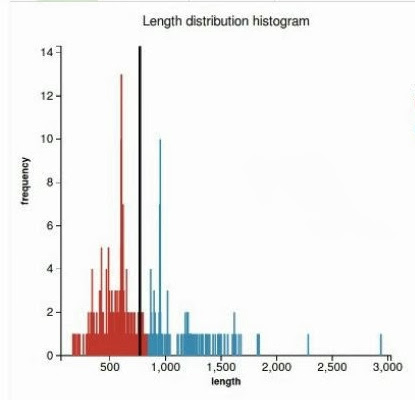
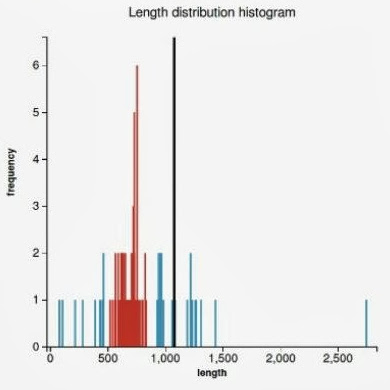
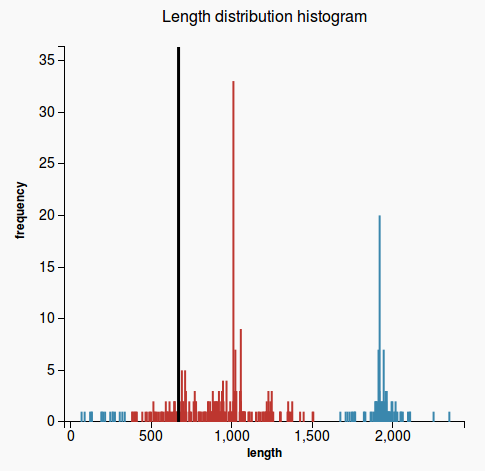
2) Length Validation via ranking
Error causes
|
Input data
|
Class information:
|
WorkflowAim: We want to see how extreme the length of the prediction is among all the other hits. Approach:
Figure 1 Figure 2 |
Codethreshold = 0.2 lengths = hits.map{ |x| x.length_protein.to_i }.sort{|a,b| a<=>b} len = lengths.length median = len % 2 == 1 ? lengths[len/2] : (lengths[len/2 - 1] + lengths[len/2]).to_f / 2 predicted_len = prediction.length_protein if hits.length == 1 msg = "" percentage = 1 else if predicted_len < median rank = lengths.find_all{|x| x < predicted_len}.length percentage = rank / (len + 0.0) msg = "TOO_SHORT" else rank = lengths.find_all{|x| x > predicted_len}.length percentage = rank / (len + 0.0) msg = "TOO_LONG" end end if percentage >= threshold msg = "" end @validation_report = LengthRankValidationOutput.new(msg, percentage.round(2)) |
PlotsNo plots are generated |
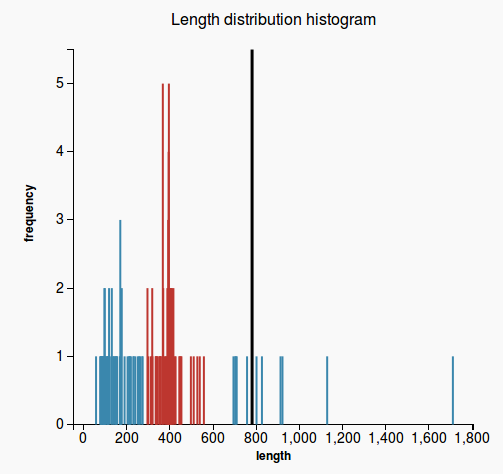
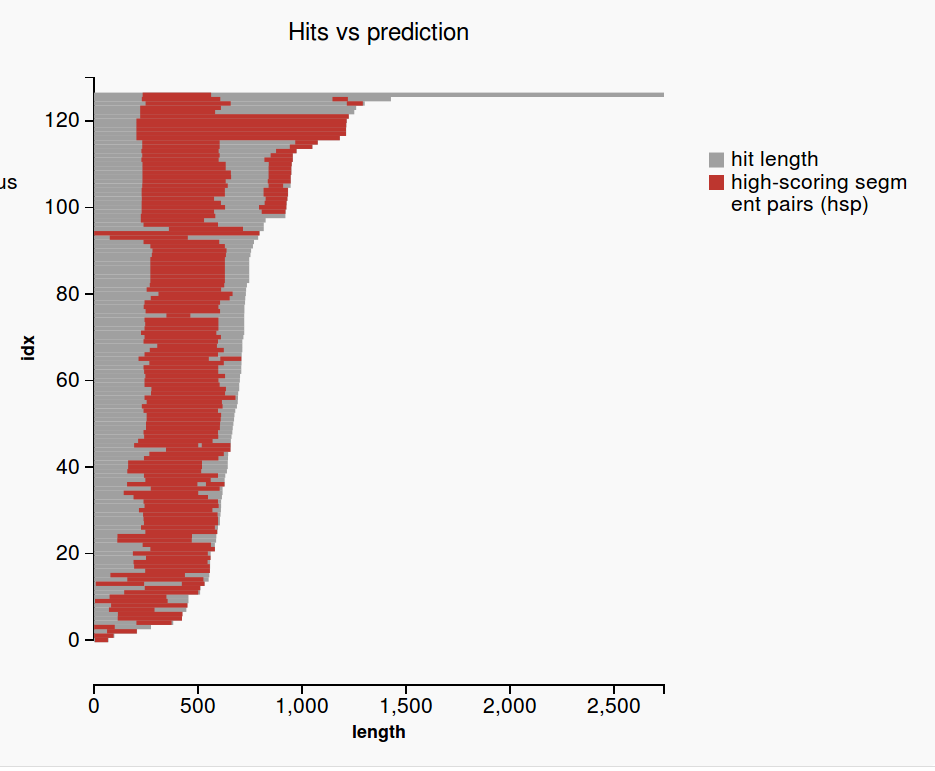
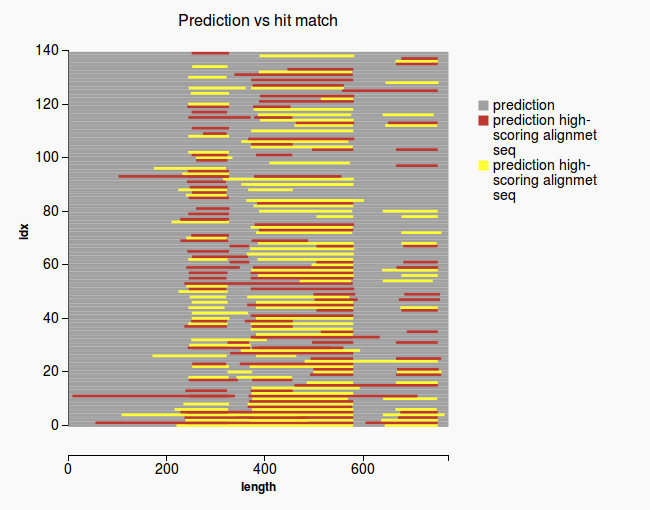
3) Duplication Check
Error causes
|
Input data
|
Class information:
|
Workflow
For each hsp:
|
Code# get the first n hits less_hits = @hits[0..[n-1,@hits.length].min] useless_hits = [] begin # get raw sequences for less_hits less_hits.map do |hit| #get gene by accession number if hit.raw_sequence == nil if hit.type == :protein hit.get_sequence_by_accession_no(hit.accession_no, "protein") else hit.get_sequence_by_accession_no(hit.accession_no, "nucleotide") end if hit.raw_sequence == "" useless_hits.push(hit) end end end rescue Exception => error raise NoInternetError end useless_hits.each{|hit| less_hits.delete(hit)} averages = [] less_hits.each do |hit| coverage = Array.new(hit.length_protein,0) hit.hsp_list.each do |hsp| # align subsequences from the hit and prediction that match (if it's the case) if hsp.hit_alignment != nil and hsp.query_alignment != nil hit_alignment = hsp.hit_alignment query_alignment = hsp.query_alignment else # indexing in blast starts from 1 hit_local = hit.raw_sequence[hsp.hit_from-1..hsp.hit_to-1] query_local = prediction.raw_sequence[hsp.match_query_from-1..hsp.match_query_to-1] # in case of nucleotide prediction sequence translate into protein # use translate with reading frame 1 because # to/from coordinates of the hsp already correspond to the # reading frame in which the prediction was read to match this hsp if @type == :nucleotide s = Bio::Sequence::NA.new(query_local) query_local = s.translate end # local alignment for hit and query seqs = [hit_local, query_local] begin options = ['--maxiterate', '1000', '--localpair', '--quiet'] mafft = Bio::MAFFT.new(@mafft_path, options) report = mafft.query_align(seqs) raw_align = report.alignment align = [] raw_align.each { |s| align.push(s.to_s) } hit_alignment = align[0] query_alignment = align[1] rescue Exception => error raise NoMafftInstallationError end end # check multiple coverage # for each hsp of the curent hit # iterate through the alignment and count the matching residues [*(0 .. hit_alignment.length-1)].each do |i| residue_hit = hit_alignment[i] residue_query = query_alignment[i] if residue_hit != ' ' and residue_hit != '+' and residue_hit != '-' if residue_hit == residue_query # indexing in blast starts from 1 idx = i + (hsp.hit_from-1) - hit_alignment[0..i].scan(/-/).length coverage[idx] += 1 end end end end overlap = coverage.reject{|x| x==0} if overlap != [] averages.push(overlap.inject(:+)/(overlap.length + 0.0)).map{|x| x.round(2)} end end # if all hsps match only one time if averages.reject{|x| x==1} == [] @validation_report = DuplicationValidationOutput.new(1) end pval = wilcox_test(averages) @validation_report = DuplicationValidationOutput.new(pval) |
PlotsNo plots are generated |
4) Gene merge Validation
Error cause
|
Input data
|
Class information:
|
Workflow
|
Codelm_slope = slope[1] y_intercept = slope[0] @validation_report = GeneMergeValidationOutput.new(lm_slope) plot1 = plot_2d_start_from(lm_slope, y_intercept) @validation_report.plot_files.push(plot1) plot2 = plot_matched_regions @validation_report.plot_files.push(plot2) @running_time = Time.now - start return @validation_report |
Plots
(1) (2)
(3) (4) |
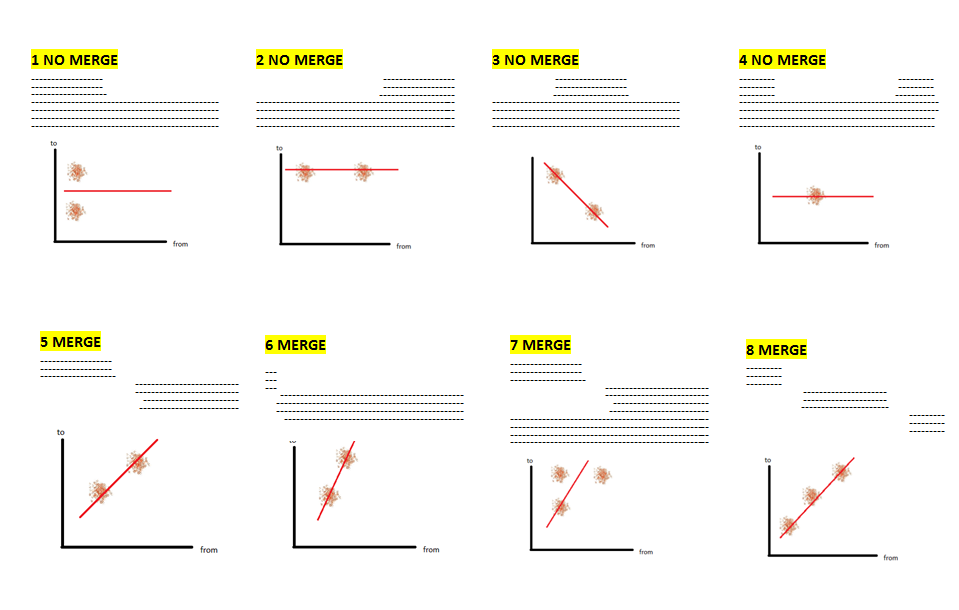
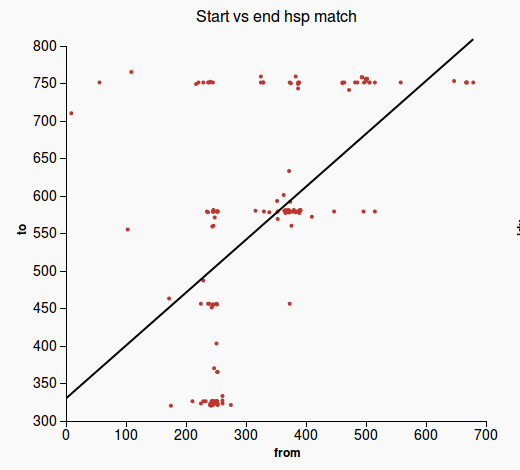
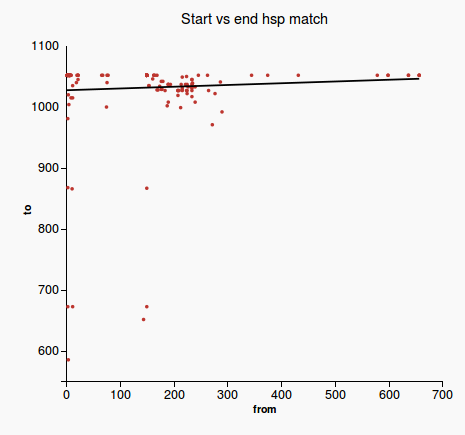
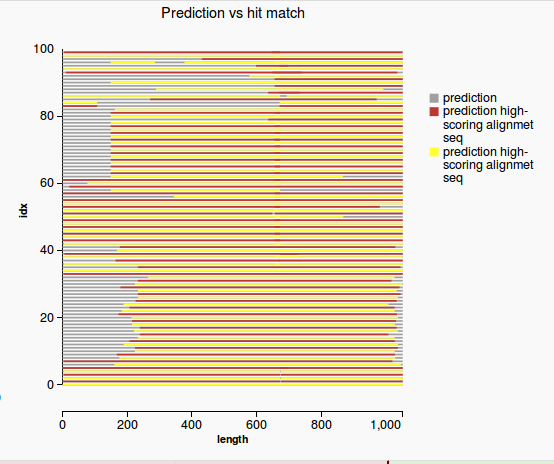
5) Validation based on multiple alignment - for proteins only
Error causes
|
Input data
|
Class information:
|
Workflow
(1) |
Code# get the first n hits less_hits = @hits[0..[n-1,@hits.length].min] useless_hits = [] begin # get raw sequences for less_hits less_hits.map do |hit| if hit.raw_sequence == nil #get gene by accession number if hit.type == :protein hit.get_sequence_by_accession_no(hit.accession_no, "protein") else hit.get_sequence_by_accession_no(hit.accession_no, "nucleotide") end if hit.raw_sequence == "" useless_hits.push(hit) end end end end useless_hits.each{|hit| less_hits.delete(hit)} begin # multiple align sequences from less_hits with the prediction multiple_align_mafft(prediction, less_hits) rescue Exception => error raise NoInternetError end sm = get_sm_pssm(@multiple_alignment[0..@multiple_alignment.length-2]) # remove isolated residues from the predicted sequence prediction_raw = remove_isolated_residues(@multiple_alignment[@multiple_alignment.length-1]) # remove isolated residues from the statistical model sm = remove_isolated_residues(sm) plot1 = plot_alignment(sm) gaps = gap_validation(prediction_raw, sm) extra_seq = extra_sequence_validation(prediction_raw, sm) consensus = consensus_validation(prediction_raw, get_consensus(@multiple_alignment[0..@multiple_alignment.length-2])) @validation_report = AlignmentValidationOutput.new(gaps, extra_seq, consensus) @validation_report.plot_files.push(plot1) |
Plots
(1) (2) |

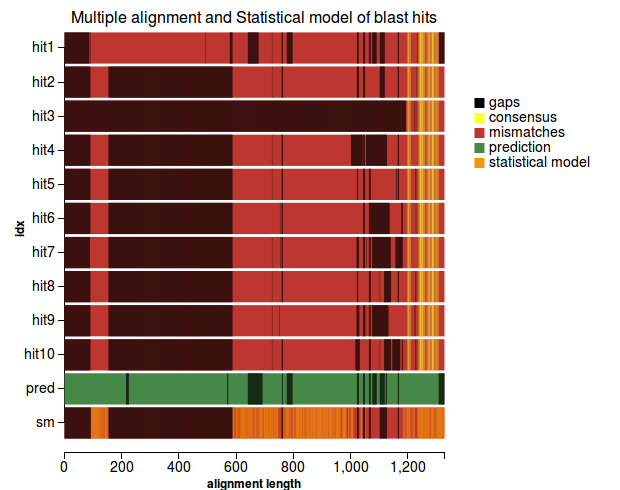
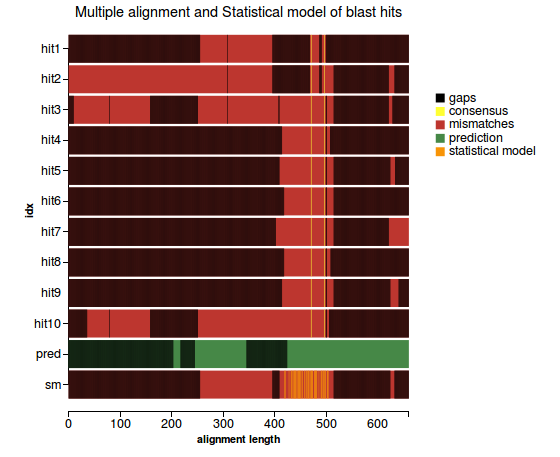
6) Blast Reading Frame Validation - for nucleotides only
Error causes
|
Input data
|
Class information:
|
Workflow
|
Codeif type.to_s != "nucleotide" @validation_report = ValidationReport.new("", :unapplicable) return @validation_report end rfs = lst.map{ |x| x.hsp_list.map{ |y| y.query_reading_frame}}.flatten frames_histo = Hash[rfs.group_by { |x| x }.map { |k, vs| [k, vs.length] }] # get the main reading frame main_rf = frames_histo.map{|k,v| v}.max @prediction.nucleotide_rf = frames_histo.select{|k,v| v==main_rf}.first.first @validation_report = BlastRFValidationOutput.new(frames_histo) |
Plots No plots are generated |
7) Open reading frame - for nucleotides only
Error causes
|
Input data
|
Class information:
|
Workflow
|
Codeif type.to_s != "nucleotide" @validation_report = ValidationReport.new("", :unapplicable) return @validation_report end orfs = get_orfs # check if longest ORF / prediction > 0.8 (ok) prediction_len = prediction.raw_sequence.length longest_orf = orfs.map{|elem| elem[1].map{|orf| orf[1]-orf[0]}}.flatten.max ratio = longest_orf/(prediction_len + 0.0) len = @prediction.raw_sequence.length f = File.open("#{@filename}_orfs.json" , "w") lst = @hits.sort{|a,b| a.length<=>b.length} f.write((orfs.each_with_index.map{|elem, i| {"y"=>elem[0], "start"=>0, "stop"=>len, "color"=>"black"}} + orfs.each_with_index.map{|elem, i| elem[1].map{|orf| {"y"=>elem[0], "start"=>orf[0], "stop"=>orf[1], "color"=>"red"}}}.flatten).to_json) f.close @plot_files.push(Plot.new("#{@filename}_orfs.json".scan(/\/([^\/]+)$/)[0][0], :lines, "Open reading frame with START codon", "", "length", "Reading Frame")) @validation_report = ORFValidationOutput.new(orfs, ratio) |
Plots
(1) |
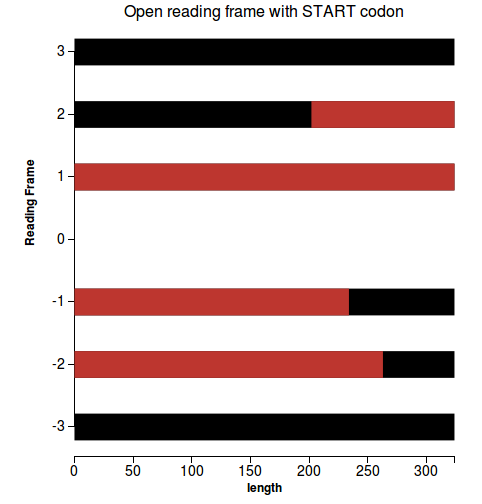
8) Codon usage - this is future work
C. Running time
Validation | 10 queries | 30 queries | Time consumers |
Length Cluster | 0.104s | 0.025s | |
Length Rank | 0.00s | 0.00s | |
Gene Merge | 0.003s | 0.01s | |
Duplication | 9.458s | 10.488s |
|
Multiple Alignment | 1.785s | 0.645s |
|
Frame Validation | 0.0s | 0.0s | |
ORF Validation | 0.015s | 0.012s |
D. Code Structure
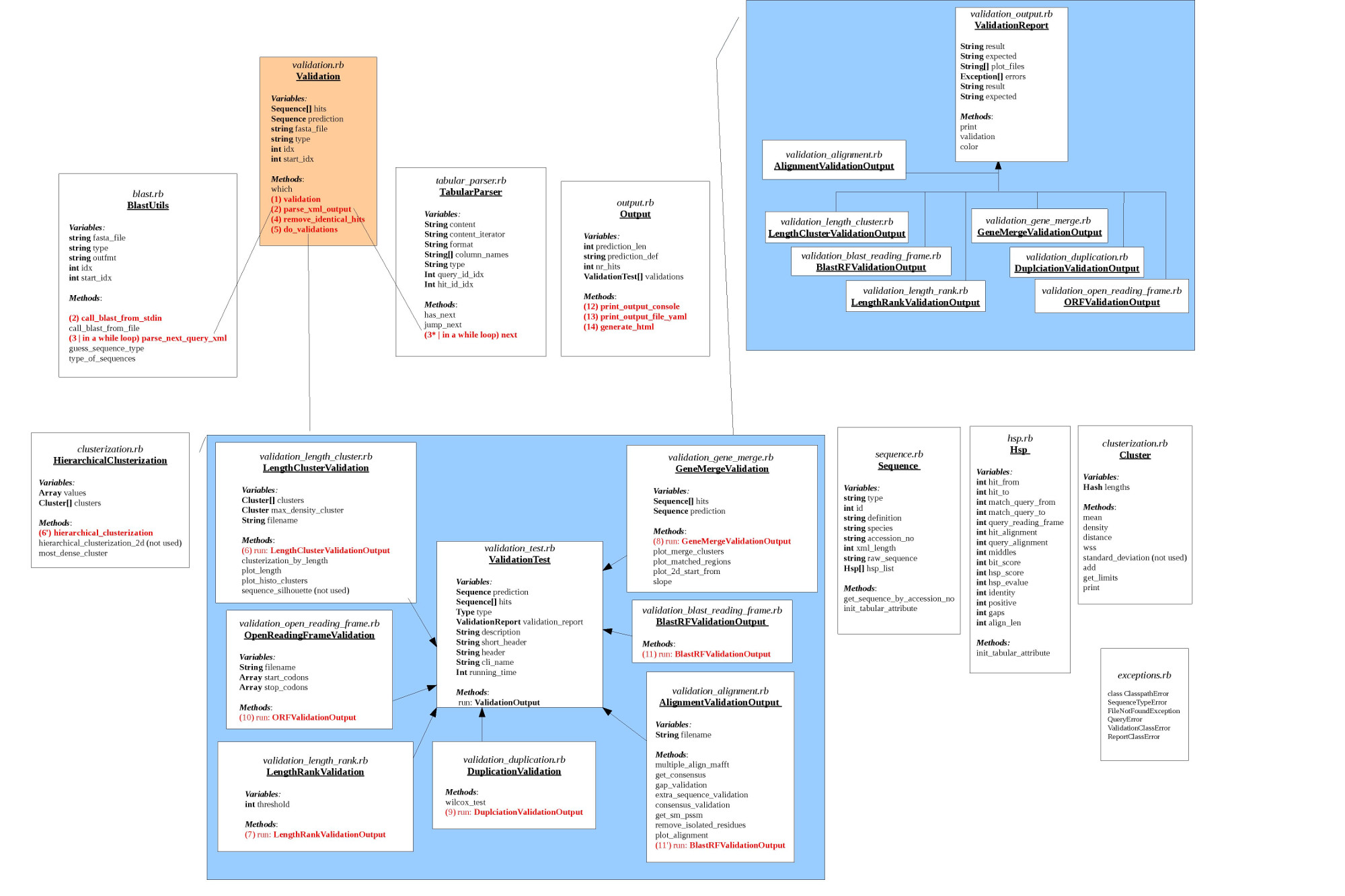
E. How to run the tool
1. Prerequisite:
- Ruby (>= 1.9.3)
- RubyGems (>= 1.3.6)
- NCBI BLAST+ (>= 2.2.25+)
- MAFFT installation (download it from : http://mafft.cbrc.jp/alignment/software/ ).
Linux and MacOS are officially supported!
2. Installation:
- Get the source code
$ git clone git@github.com:monicadragan/gene_prediction.git
- Be sudo and build the gem
$ sudo rake
- Run GeneValidation
$ genevalidator [validations] [skip_blast] [start] [tabular] [mafft] [raw_seq] FILE
Example that runs all validations on a set of ant gene predictions:
$ genevalidator -x data/prot_Solenopsis.xml data/prot_Solenopsis.fa
To learn more:
$ genevalidator -h
3. Input
- FASTA file with the prediction queries (queries must be of the same type: mRNA or proteins. Mixed type FASTA files are not allowed!).
- [optional] Precalculated BLAST output file in xml format (-outfmt 5) or tabular format (-outfmt 6 or -outfmt 7 or “Hit Table (CSV)” in the online version of BLAST). Please specify the --tabular argument if you use tabular format input with nonstandard columns. Also, we search only in protein database using a blastp for protein queries and blastx for nucleotide queries.
4. Output
By running GeneValidator on your dataset you get numbers and plots. Some relevant files will be generated at the same path with the input file. The results are available in 3 formats:
- console table output
- validation results in YAML format (the YAML file has the same name with the input file + YAML extension)
- html output with plot visualization (the useful files will be generated in the 'html' directory, at the same path with the input file). Check the index.html file to see the output.
! Note: for the moment check the html output with Firefox browser only !
(because our d3js cannot be visualized from other browsers without a local webserver)
5. Scenarios
5.1 Quickly test the tool or validate a small set of predictions (internet connection required).
$ genevalidator data/prot_Solenopsis.fa
What happens backstage: blast hits and raw sequences are retrieved from remote databases and analysed for each query.
5.2 Big input files (>100 queries) & you have access to a more powerful machine that pre-calculates overnight the blast output file. Use blastp for proteins and blastx for nucleotide queries.
5.2.1 If you have time and a really powerful machine you can take the blast xml output. This way you will save time when running GeneValidator (because you precalculate local alignments with BLAST)
Example:
# take blast output for protein queries (for mrna queries use blastx)
$ blastp -query predictions.fasta -db /path/to/db/nr/nr -outfmt 5 -max_target_seqs 200 -num_threads 48 -out predictions_blast.xml
# run validations
$ genevalidator -x predictions_blast.xml predictions.fasta
5.2.2 Take blast tabular output (customized so that you have all the necessary columns -- see the table with data needed for validations in section B)
Example:
# take blast output for protein queries
$ blastp -query predictions.fasta -db /path/to/db/nr/nr -outfmt "6 qseqid sseqid sacc slen qstart qend sstart send length qframe pident evalue" -max_target_seqs 200 -num_threads 48 -out predictions_blast.tab
# run validations
$ genevalidator -x predictions_blast.tab -t "qseqid sseqid sacc slen qstart qend sstart send length qframe pident evalue" predictions.fasta
5.3 You are interested in certain validations only (let’s take length validations and duplication check). Look up the CLI names for these validations in the table (Section B) and add them as argument so that GeneValidator will process those 3 only.
$ genevalidator -x predictions_blast.xml predictions.fasta -v ‘lenc lenr dup’
5.4 Start validating from a certain query in the fasta file: the following command skips the first 9 queries
$ genevalidator -x predictions_blast.xml predictions.fasta -s 10
F. Add a new validation
The code architecture is designed so that it is very easy to add new validations. There is a ‘template’ that the code of the new validation must fit, which is discussed further:
Extend 2 parent classes
Initialize internal variables (which initially have some default values):
Initialize internal variables (which initially have some default values):
|
Add the new validation to the toolAdd the validation to the validations list, which is further processed for yaml/html/console visualization # context code # validations = [] validations.push LengthClusterValidation.new(@type, prediction, hits, plot_path, plots) … ## what you need to add is this line that adds your validation class to the list of validations. Your class must extend ValidationTest ## validations.push YourValidationClass(type, prediction, hits, other_arguments) # context code # # check the class type of the elements in the list # this will raise an error if YourValidationClass does not extend ValidationTest validations.map do |v| raise ValidationClassError unless v.is_a? ValidationTest end # run validations validations.map{|v| v.run} # check the class type of the validation reports # this will raise an error if the run method of YourValidationClass does not return ValidationReport validations.map do |v| raise ValidationClassError unless v.validation_report.is_a? ValidationReport end |
Add plotsA. Generate json file according to the type of plot1) (clustered) bars plot, main cluster is in red: [ [{"key":109,"value":1,"main":false}], [{"key":122,"value":32,"main":true},{"key":123,"value":33,"main":true}], [...]] 2) scatter plot: [{"x":1,"y":626},{"x":1,"y":626}...] 3) lines: [{"y":1,"start":0,"stop":356,"color":"black"},{"y":1,"start":40,"stop":200,"color":"red"}...] B. Create a plot object, according to the plot typeplot1 = Plot.new(filemane, plot_type # which can be :scatter, :line or :bar plot_title, legend**, xTitle, yTitle, aux_parameter1*, aux_parameter2*) ** legend is a String that _must_ have the following format: "item, color1;item with spaces, color2" * auxiliary parameters were used by now for passing the length of prediction (in length histogram) or the slope of the regression line (gene merge validation) or the number of lines (line plots) C. Add the plot object to the plot_files array@validation_report.plot_files.push(plot1) |
G. Output results
Some validation outputs for protein and mRNA are available here:
http://swarm.cs.pub.ro/~mdragan/gsoc2013/genevalidator