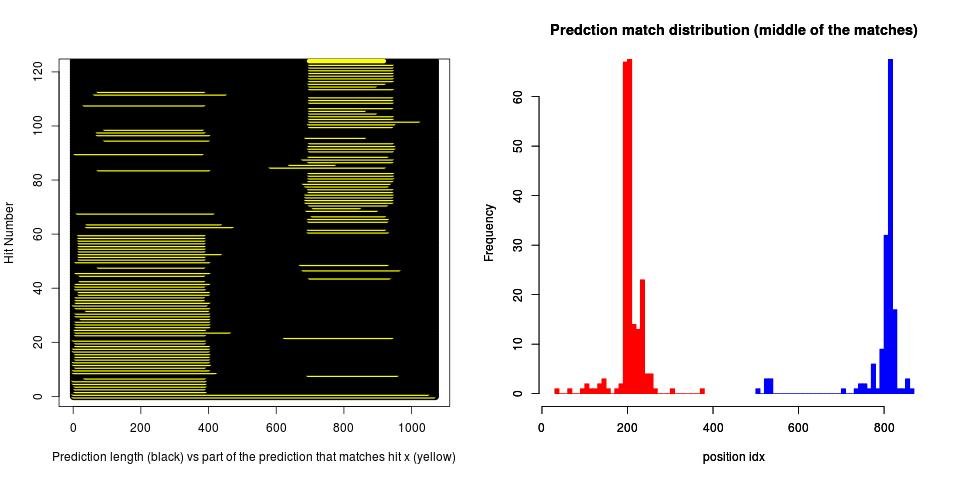
By now we validate 4 things about the predicted genes: the length (by clusterization and ranking - the rank of the prediction among all the hits), the reading frame, whether there is a duplication in the prediction and whether the prediction is in fact a merge between multiple genes.
We adjusted our previous approach for merge detection in order to have fewer false positives. I'll briefly explain how we do the validation now:
- we plot a 2D graph by using the start/end offsets of the matched regions in the prediction on the two axis
- we draw a line obtained by linear regression
- a gene merge is present if the slope of the line is between 0.4 and 1.2
- these thresholds were chosen empirically and will be adjusted after analyzing a larger amount of data.
Here it is a simplified drawing explaining this:

The output can be provided in html format (if you use the proper command line arguments). Here you can find sume examples, grouped by validation test:
Length validation: http://swarm.cs.pub.ro/~mdragan/gsoc2013/solenopsis_length_test/
Gene merge validation: http://swarm.cs.pub.ro/~mdragan/gsoc2013/one_direction_gene_merge/
Duplication check: http://swarm.cs.pub.ro/~mdragan/gsoc2013/duplications/
If I convinced you, try the application yourself:
Update: please clone from the developmenet branch